Metagenomics: A New Way To Study Microbial Sciences

Jun 13, 2019 • 66 views
Introduction
Metagenomics a growing field of environmental genomics. Metagenomics is the total genetic complement of the cells present in a particular environment.
So the total genome content of a microbial community is known as metagenomics.
New genomics techniques offer culture-independent approaches to studying microbial community.
Around ten years later, in 1998, the term “metagenome” appeared, when Handelsman and collaborators described the importance of soil microorganisms as sources for new natural compounds.
To do this DNA fragments are extracted directly from the environment and cloned into plasmids vectors. Then the library of the environmental DNA fragments can be made and amplified.
A stable source of the nucleotide sequence is generated that reflects the diversity of microbead growing in nature, not just those that can be grown in the laboratory.
The nucleotides are then sequenced and analyzed, or expressed in a microbial host and screened for a specific function, such as the production of novel antimicrobial compounds.
One field that metagenomics has revolutionized is marine microbiology.
At the beginning of the metagenomic studies, the use of Sanger sequencing technology provided important progress in the field. However, the advent of next-generation sequencing (NGS) technologies capable of sequencing millions of DNA fragments simultaneously, at a low cost, greatly bolstered the field. Comparatively, NGS platforms can recover up to 5000 Mb of DNA sequence per day with costs at about 0.50$/Mb, while Sanger sequencing methodology allows about 6 Mb of DNA sequence to be created per day with costs at about 1000 higher.
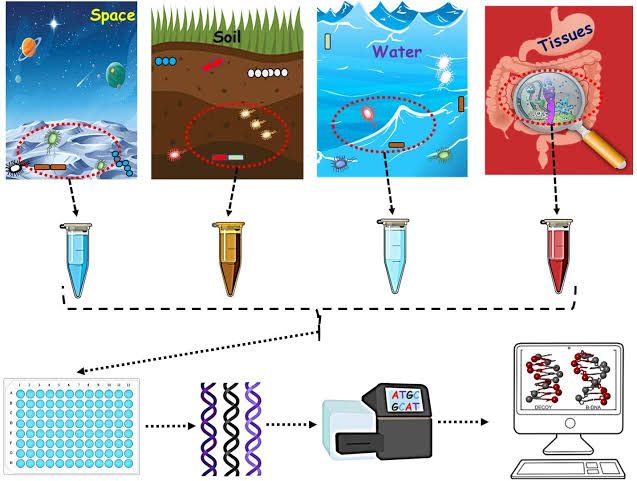
One case study of Sargasso sea was performed by J. Craig vendor, Hamilton Smith, and colleagues. They determined the bacterial and archaeal biodiversity of the Sargasso Sea, the portion of the Atlantic ocean that surrounds Bermuda. They collected seawater and used filtration to excluded viruses and eukaryotes. An environmental genomic library was prepared from extracted DNA. At least 1800 phylotypes were represented, among which 145 phylotypes were previously unknown and most likely presents new species.
Phylogenetic diversity was further evaluated by PCR amplification of genes, such as the recombine recA, the 16S rRNA gene, and the gene that encodes RNA polymerase B(rpoB). These sequences are highly conserved, making them good candidates for assessing species diversity.
They reported the discovery of 1.2 million previously unknown genes, including over new 700 new proteorhodopsin-like photoreceptors from the taxa not previously known to possess light-harvesting capabilities.
Review of literature
The human gut microbiota makes fundamental contributions to host metabolism and immune system. Therefore, perturbations in its composition, a process known as dysbiosis, have an important role in the development of several chronicle diseases, mainly intestinal inflammatory disorders. The human microbiome represents all the genes present in the human genome and those present in the trillions of microbes living in and on adults.
The microbiome is at least several hundred times larger than our own and encodes a variety of metabolic processes we lack, such as synthesis of vitamin K and the degradation of a variety of otherwise indigestible food items.
A major portion of these mutualistic microbes is found in the human gut, where they occupy niches that make contributions to nutrient processing, pathogen colonization resistance, and mucosal immune system development.
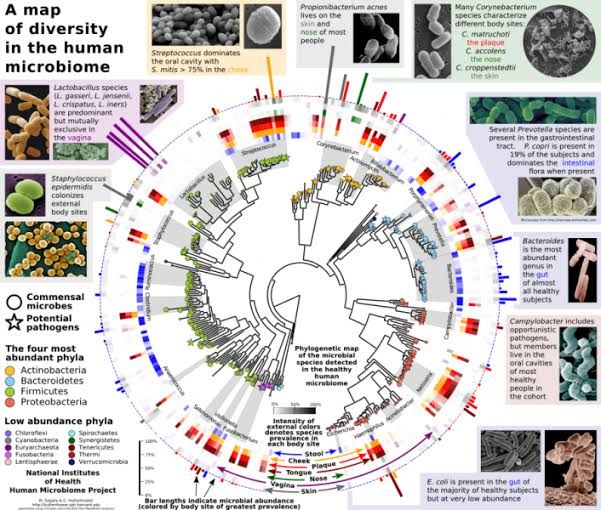
Most of the organism inhabits large intestine which belongs to one of the two phylogenetic groups of bacteria, the bacteroids, and the Firmicutes. A fascinating discovery was that gut microbiome is related to obesity in human and mouse model.
According to the finding, it was because of fermentation firmicutes species which converts more dietary fiber into fatty acids that can be absorbed by the host.
Culture-independent methods, in particularly next-generation sequencing technologies, have prompted a huge breakthrough in our knowledge regarding the microbial communities colonizing the human body and their functional beneficence to host health.
Bacterial identification using 16S rRNA sequencing
New generation sequencing technologies are capable of processing high amount of DNA in a relative short time using 16S ribosomal (16S rRNA) genetic information. Several high throughput platforms such as 454 Roche GS FLX, Applied Biosystems SOLiD System, Illumina HiSeq and MiSeq System and Ion Torrent Personal Genome Machine (PGM) have been used for this kind of metagenomic approach.
PCR-amplified small sequences of 16S rRNA gene from extracted DNA, generally using universal primers annealing conserved nucleotides to amplify one or more fragments of variable regions.
The sequences at a pre-defined level of identity stand for grouped clusters of similar sequencing reads, known as Operational Taxonomic Unit (OTU), which corresponds to a group of very similar 16S sequences. Reference databases (GreenGenes, myRDP, NCBI) are used to classify OTUs providing identification of taxonomy, relative frequencies and diversity of community composition in samples obtained from the certain ecosystem.This approach allows identification of new species and investigation of low-abundance bacteria and even uncultivated gut microbial communities from a single analysis. In addition, these technologies are faster and more accurate compared to classical identification methods (cloning and culture).
Shotgun sequencing
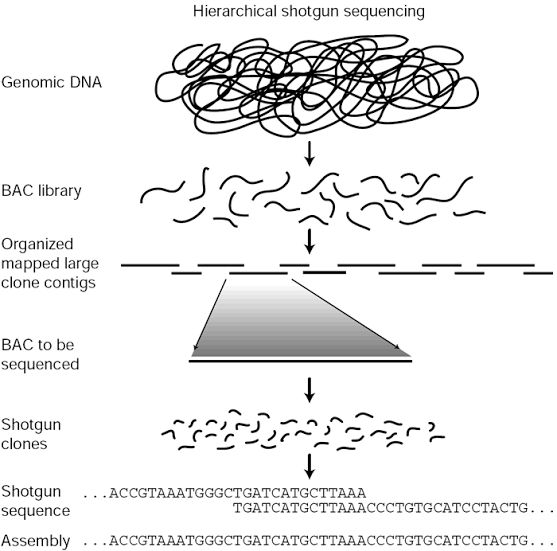
The whole metagenome sequencing can be performed using a shotgun approach. In such genomic survey strategy, multiple continuous overlapping sequences (contigs), which are assembled from fragmented sequences and obtained from total purified genomic DNA, are used for identifying genes through alignment with bacterial reference genomes and databases (KEGG, SEED and NCBI).
Shotgun approach is quite versatile, in which the samples can be submitted to various methods, including nebulization, endonucleases, or sonication for random fragmentation of DNA, sequencing a subsequent contig assembly and annotation. Furthermore, advanced computational methods applying different algorithms are frequently being developed for more accurate assembly and annotation of genes, thus allowing functional characterization in complex environments like the human gut.
Conclusion
It concludes that human microbiome needs an extensive characterization to understand the mutualistics roles of microbes in different metabolic pathways and to understand the pathogencity mechanism of bacteria.In this context, further advances in the area are required to achieve a more precise definition of gut microbiome role, which could allow investments for the search of more effective therapies.
Recommended

Kasam Ki Kasam Chords - A.R.Rahman & Shaan - Kabhi Khushi Kabhi Gham
Mera Yaar Chords - Javed Bashir & Shankar Ehsaan - Bhaag Milkha Bhaag